|
|
|
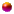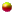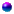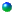  Studies Towards the Synthesis of
Phomactin D. A synthesis of the natural product Phomactin D 205
is being pursued in our group where the key step will be an asymmetric
cyclohexadienone annulation of Fischer carbene complexes.
Phomactin D was identified in a screen for platelet activating
factor (PAF) antagonist activity that Schering Plough and Sankyo have
independently made over the last decade on the marine fungus Phoma
sp.. Both groups have
discovered active compounds which possess the same rare cleomane carbon
skeleton. The most active
compound is phomactin D 205
which has an IC50 for platelet aggregation of 0.80 uM.
The only total synthesis of Phomactin D reported to date was
accomplished in 36 steps from L-ascorbic acid.[3] Studies Towards the Synthesis of
Phomactin D. A synthesis of the natural product Phomactin D 205
is being pursued in our group where the key step will be an asymmetric
cyclohexadienone annulation of Fischer carbene complexes.
Phomactin D was identified in a screen for platelet activating
factor (PAF) antagonist activity that Schering Plough and Sankyo have
independently made over the last decade on the marine fungus Phoma
sp.. Both groups have
discovered active compounds which possess the same rare cleomane carbon
skeleton. The most active
compound is phomactin D 205
which has an IC50 for platelet aggregation of 0.80 uM.
The only total synthesis of Phomactin D reported to date was
accomplished in 36 steps from L-ascorbic acid.[3]
The
key step in the synthesis of phomactin D is the cyclohexadienone
annulation with a 1,4-asymmetric induction.
The original study of this process for 1,4-asymmetric induction in
the construction of a cyclohexadienone were performed on the simple
complex 60 and the chiral propargyl ether 40 as was discussed in Scheme VI.[4]
In this study it was found that the reaction was both
stereoselective and stereospecific.
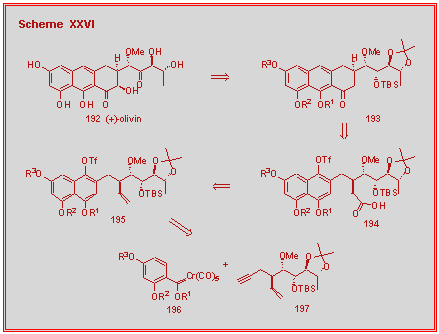
Our retrosynthesis of phomactin D is shown in Scheme XXVIII and
involves the ring closing methathesis of intermediate 210 to give the [9.3.1]-bicylopentadecanone 209. The
stereoselective synthesis of the cyclohexadienone 210
was achieved with a 94 : 6 selectivity from the reaction of the carbene
complex 211 with the chiral
propargyl ether 212.
This selectivity could be improved to 98 : 2 if the temperature
was reduced to 55 oC. The
chiral center of the propargyl ether does not appear in the natural
product since oxidation to a ketone will need to occur at some point in
the synthesis. Nonetheless,
the chiral propargyl ether chiral center will be used, via the
cyclohexadienone annulation, to set the correct absolute configuration
of the chiral center in the six-membered ring of the natural product.
Studies directed to the completion of the synthesis of phomactin
D are now underway.
|
|
Synthesis
of Members of the Colchicine Family. (-)-Colchicine 213 (Scheme
XXIX), the major alkaloid from Colchicum
autumnale, is one of the oldest known natural products. It binds to
the cytoskeletal protein tubulin, disrupting the microtubule-dependent
functions in the cell and thereby suppressing the cell division process.
(-)-Allocolchicine 222,
is also a natural product and is an isomer of colchicine and has similar
biological activity to colchicine.
Allocolchicine has the tropone ring replaced by an arene ring.
Only natural (-)-7S-colchicine 213, which
adopts an aR biaryl
configuration, but not its 7R
enantiomer, binds effectively to tubulin.
We are presently pursuing a total synthesis of (-)-colchicine 213, and several of its analogs by a method which relies on the
reaction of a carbene complex of the type 130
in Scheme XIII where there is a central to axial chirality transfer
during the key benzannulation step.[5]
We
have also been interested in the synthesis of other natural products in
the colchicine family and we have recently completed the first total
synthesis of (-)-allocolchicine 222
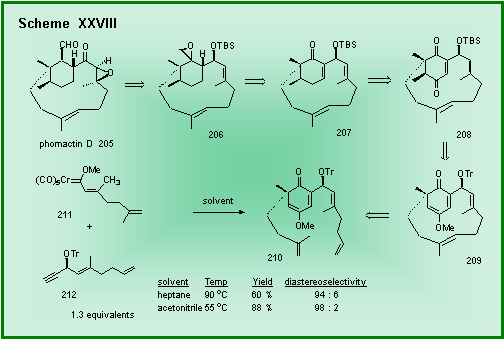
which is outlined in Scheme XXIX.[6]
The key step is the Diels-Alder reaction of the diene 217
with methyl propriolate. This
reaction was found to go at 110 oC to give, after
aromatization with DDQ, a single regioisomer of the allocolchicinoid 218.
The TBS protected alcohol function in 217
will later become the nitrogen substituent in allocolchicine and is also
crucial to the regioselectivity of the Diels-Alder reaction.
In the absence of this substituent, the Diels-Alder reaction
proceeds to give a nearly equal mixture of cycloadducts as indicated in
the formation of 215.
Asymmetry is introduced during the reduction of the ketone 219.
This is accomplished by the recent method of Singram and
coworkers who found that ketones could be reduced with high inductions
with lithium borohydride in the presence of the chiral Lewis acid 216.[7]
Applied to 219, this method gives the alcohol 220 in 91 % ee. The
conversion of 220 to (-)-allocolchicine
occurred with no substantial loss of optical purity in three steps.
A final crystallization gave the optically pure natural product.
|
|
Studies
on the Synthesis of Fostriecin. Fostriecin (CI-920) 223,
is an interesting phosphate ester which has antitumor activity and has
been brought to phase I clinical trials.
It has recently been shown by Roberge in British Columbia that it
is an inhibitor of protein phosphatases 1 and 2A, a property not
previously described for an antitumor drug.[8]
The antitumor activity has been shown to be the result of an
interaction with topoisomerase II, but its protein phosphatase activity
is involved in its ability to inhibit the mitotic entry checkpoint.
This means it induces cells to enter mitosis.
Thus it is suspected that it is a tumor promoter and this is
apparently being checked by a number of research groups.
Fostriecin is somewhat unstable and when used in the clinic must
be stored frozen in buffer. This
instability is presumably due to the trienediol portion of the molecule.
In addition to the total synthesis of fostriecin, our group is
involved in the synthesis of analogs that would hopefully be more stable
and yet still retain activity.
|
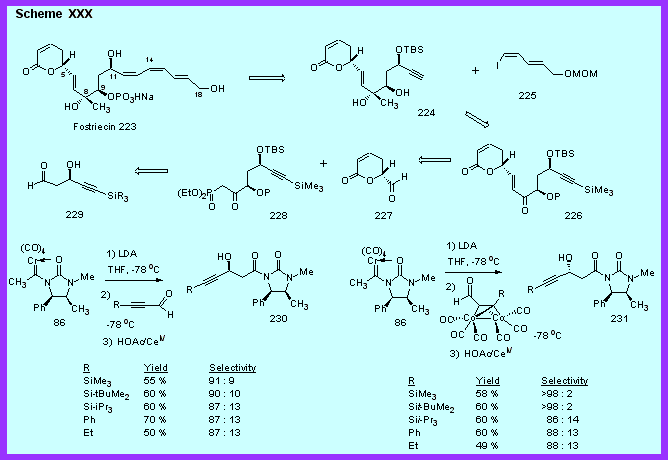
Our synthetic strategy for the synthesis of fostriecin is shown in Scheme
XXX and involves the introduction of the sensitive trans, cis, cis-triene
portion along with introduction of the phosphate on the C-9 second
alcohol at the end of the synthesis.
A second key disconnection is the Horner-Wadsworth-Emmons
reaction that couples the lactone unit 227
with the central fragment 228
which in turn is to be derived from the b-hydroxy aldehyde 229.
The strategy outlined in Scheme XXX is a result of the
ability of chiral imidazolidinone carbene complexes to stereoselectively
provide asymmetric aldol reactions with 2-alkynals. In
previous work we have shown that the chiral imidazolidinone carbene
complex 86 is an excellent
chiral acetate enolate equivalent (See section
IV on aldol reactions).[9]
A minimum of 20 : 1 selectivity can be achieved with aryl
aldehydes, branched aliphatic aldehydes and unbranched aliphatic
aldehydes. These carbene
complexes can be prepared in good yields and in a single step.
The chemical yields of the aldol reactions for simple aldehydes
are in the 80 - 90 % range, which includes removal of the metal in the
workup by stirring the crude reaction mixture for a few minutes with
aqueous cerium (IV).
Given the small size of an
acetylene group, it was quite surprising to find that the aldol
reactions of carbene complex 86
with alkynals occur with very good selectivities as indicated by the
data in Scheme XXX. Even
more surprising was the fact that they occur with the opposite
selectivity that is observed for simple alkyl aldehydes.
This selectivity can be reversed by employing dicobalt complexed
alkynals which give the aldol adducts 231.
The stereoselectivity of these reactions was confirmed by an
X-ray structure of 231 (R =
TBS) in collaboration with Arnold Rheingold. The cobalt complexed alkynals thus give the
"normal" selectivity. While
the source of this selectivity reversal for alkynals is unknown at this
time, our current efforts are directed to the completion of the
synthesis of fostriecin.
|
|
Studies on the Synthesis of Taxol. Taxol is a clinically important agent for the treatment of
ovarian and other cancers and is the number one selling cancer drug in
the world. Our preliminary
studies reveal that the reaction of Fischer carbene complexes with
enynes has the potential to provide for rapid access to the taxol
skeleton. As described in Section
VI on cyclobutanone formation, the reaction of Fischer carbene
complexes with enynes can be controlled by the substitution pattern on
the alkene to give either bicyclo-[3.2.0]-heptanones or bicyclo-[3.1.1]-heptanones.[10]
Subsequently, we have discovered that the reaction to give the
latter can be utilized to provide a fundamentally new strategy for the
synthesis of taxol and related molecules and this is indicated in Scheme
XXXI. 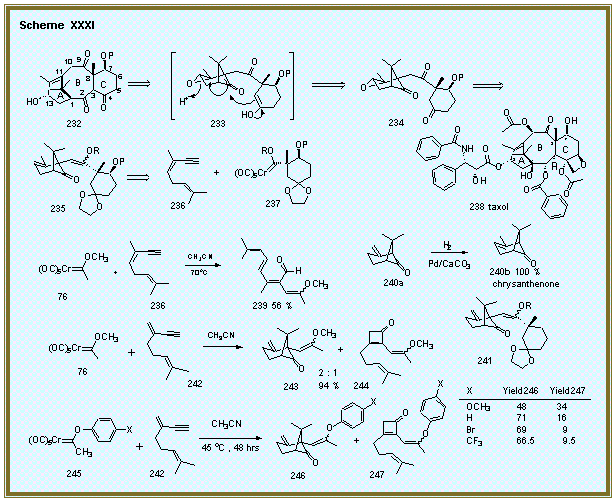
We
have published our initial studies on the viability of the reaction of
Fischer carbene complexes with enynes as a key tactic in a strategy for
the synthesis of taxol and taxol analogs.[11]
The first generation strategy involved the access of the
tricyclic intermediate 235
from the reaction of the enyne 236
and the carbene complex 237.
This strategy was based on the reaction of complex 76
with enyne 112 which gave the
bicyclo [3.1.1] heptanone 114
in 69 % overall yield (See Section
VI). The strategy was
adapted to include the extra alkene in enyne 236 which, after reaction with complex 237 and epoxidation, would give intermediate 234. The plan was to
construct the eight membered ring and introduce the bridgehead
double-bond by a Grob fragmentation of the epoxide.
This was not to be as it was found that the reaction of the enyne
236 with the complex 76
did not give a bicyclic species, but rather the highly unsaturated
aldehyde 239.
A mechanistic interpretation of this outcome suggested a second
generation strategy which involved the reaction of the exocyclic
methylene substituted enyne 242
with complex 237 to give the
bicyclic species 241.
This would then be followed by the isomerization of the exo-cyclic
double-bond in 241 to give the intermediate 235
in the original strategy. The
possibility for this transformation is suggested by the isomerization of
the bicycloheptanone 240a to 240b
in quantitative yield.[12]
The reaction of complex 76 with enyne 242 was
performed as a model study for the second-generation strategy.
Although this reaction gave high yield, it unfortunately gave a
mixture of the desired product 243 along with the cyclobutenone product 244. It was found that
the ratio of these products could be controlled to some extent by
electronic tuning of the carbene complex.[13]
Electron poor phenoxy complexes of the type 245
lead to increased proportions of the desired product 246
as indicated by the data in Scheme 1.
Further elaboration of the second-generation strategy for the
synthesis of taxol is under investigation.
|
|
Synthesis of Chloroamphenicol.
One of the oldest anti-bacterial agents is chloroamphenicol which
was first isolated from Streptomyces Venezuelae in 1947.
This antibiotic is obtained commercially by chemical synthesis
and is biological active only as its 2R,3R enantiomer. It is used clinically as a broad spectrum antibiotic and is
particularly useful for the treatment of salmonella,
typhi, rickettsia and meningeal
infections. As a result of
its link to bone marrow depression, its use is reserved for serious
infections with organisms which have been demonstrated to be resistant
to all other appropriate anti-microbial agents.
A number of chemical syntheses of racemic chloramphenicol have
been reported as well as a few in the last decade that are selective for
the formation of (-)-chloramphenicol.
We have recently published an asymmetric synthesis of optically
pure (-)-chloramphenicol that is the shortest synthesis of the
enantiomerically pure antibiotic to date.[14]
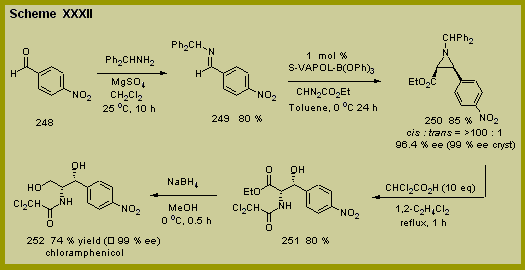
|
|
The
synthesis of (-)-chlormaphenicol is presented in Scheme XXXII and
features an asymmetric catalytic aziridination reaction that has
recently been developed in our laboratories (See
Section IV).[15]
The synthesis begins with commercially available para-nitrobenzaldehyde
and its conversion to its corresponding benzydyl imine 249 which is obtained in 85 % yield after crystallization from
ethanol. The aziridination
is carried out with 1 mole percent of the catalyst prepared from the
VAPOL ligand in toluene at 0 oC to give the aziridine 250 in 80 % yield, 96 % ee and with a >100:1 cis/trans selectivity.
The enantiomeric purity of this aziridine could be improved to 99
% ee with a single crystallization from hexane / methylene chloride (84
 %
yield, 1st crop). Treatment
of the optically pure aziridine 250
with 10 equivalents of dichloroacetic acid in refluxing
1,2-dichloroethane for one hour gave the hydroxy acetamide 251
in 80 % yield as a single diastereomer.
Completion of the synthesis was accomplished by reduction of the
ethyl ester with sodium borohydride which gave (-)-chloroamphenicol in
74 % yield and in greater than 99 % enantiomeric excess.
The synthesis of enantiomerically pure (-)-chloroamphenicol was
thus achieved in four steps from commercially available starting
materials in 38 % overall yield. %
yield, 1st crop). Treatment
of the optically pure aziridine 250
with 10 equivalents of dichloroacetic acid in refluxing
1,2-dichloroethane for one hour gave the hydroxy acetamide 251
in 80 % yield as a single diastereomer.
Completion of the synthesis was accomplished by reduction of the
ethyl ester with sodium borohydride which gave (-)-chloroamphenicol in
74 % yield and in greater than 99 % enantiomeric excess.
The synthesis of enantiomerically pure (-)-chloroamphenicol was
thus achieved in four steps from commercially available starting
materials in 38 % overall yield.
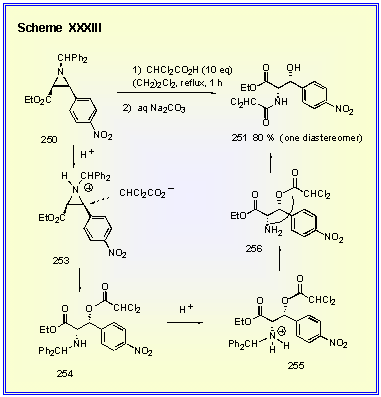
The
conversion of 250 to 251 actually involves three steps in one as outlined in Scheme
XXXIII. In the first
step, dichloroacetic acid protonates the aziridine nitrogen which is
then opened by its counter ion with inversion to give the bis-ester 254.
A second molecule of dichloroacetic acid then protonates the
amine in 254 which leads to
cleavage of the benzhydryl group and the formation of the free amine 256. The final step is an intramolecular acyl transfer which is
thermodynamically driven to give the amide 251.
|
[1] Gilbert, A. M.; Miller, R.;
Wulff, W. D., Tetrahedron,
1999, 55, 1607.
[2] Liptak, V. P.; Wulff, W.
D., Tetrahedron, 2000,
56, 10229.
[3] Miyaoka, H.; Saka, Y.;
Miura, S.; Yamada, Y.; Tetrahedron
Lett., 1996, 37,
7107.
[4] Hsung, R. P.; Quinn, J.
F.; Weisenberg, B. A.; Wulff, W. D.; Yap, G. P. A.; Rheingold, A.
L., J. Chem. Soc., Chem. Commun.,
1997, 615.
[5] Vorogushin, A. V.; Wulff,
W. D.; Hansen, H.-J., J. Am.
Chem. Soc., 2002, 124,
6512.
[6] Vorogushin, A. V.;
Predeus, A. V.; Wulff, W. D.; Hansen, J.-J., J. Org. Chem., 2003, 68,
0000.
[7] Suri, J. T.; Vu., T.;
Hernandez, A.; Congdon, J.; Singram, B., Tetrahedron Lett., 2002, 43,
3649.
[8] Ho, D. T.;
Roberge, M.; Carcinogenesis 1996, 17,
967
[
[
[9] Powers, T. S.; Shi, Y.;
Wilson, K. J.; Wulff, W. D., J.
Org. Chem., 1994, 59,
6882.
[10] Kim, O.K.; Wulff, W. D.;
Jiang, W.; Ball, R. G., J.
Org. Chem., 1993, 58,
5571.
[11] Jiang, W.; Fuertes, M. J.;
Wulff, W. D., Tetrahedron,
2000, 56, 2183.
[12] Kulkarni, Y. S.; Snider,
B. B., J. Org. Chem., 1985,
58, 5571.
[13] Jiang, W.; Fuertes, M.
J.; Wulff, W. D., Tetrahedron,
2000, 56, 2183.
[14] Loncaric, C.; Wulff, W.
D.; Org. Lett., 2001,
3, 3675.
[15] Antilla, J.; Wulff, W. D.,
Angew. Chem. Int. Ed. Engl.,
2000, 39,
4518.
|
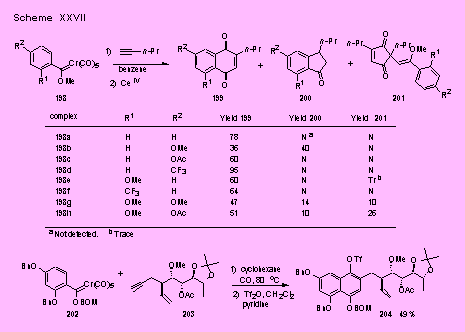 trifluoromethyl
substituted complex gives a 95 % yield of the quinone.
trifluoromethyl
substituted complex gives a 95 % yield of the quinone.