|
Carbene
Complexes
Introduction
I. Benzannulation
Reaction
II.
Cyclohexadienone
Annulation
III. Tautomer
Arrested Annulation
IV. Aldol
Reaction
V. Diels-Alder
Reaction
VI. Cyclobutanone
Formation
VII. Biaryl Synthesis
VIII.
Macrocycles
Asymmetric
Catalysis
I. Ligand Design
and
Synthesis
II. Asymmetric
Diels-Alder
Reaction
III. Imino Aldol
Reaction
IV. Asymmetric
Aziridination
Synthesis of Natural Products
and Pharmaceuticals
|
|
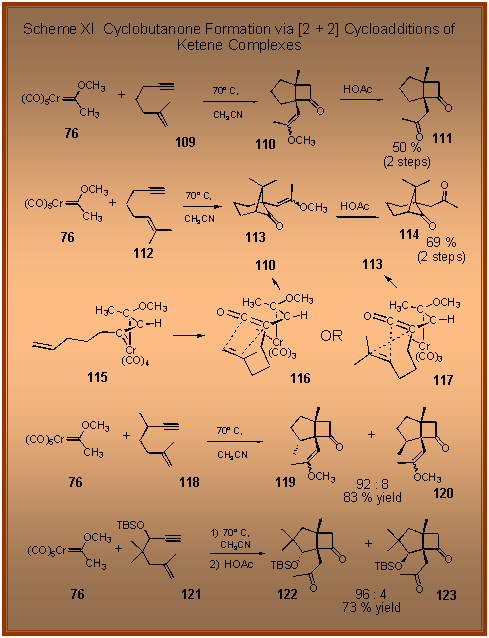
The
reactions of Fischer carbene complexes with enynes were
initially carried out to probe whether the carbene complex would
react first with the alkyne or with the alkene.
We found that the alkyne function was much more reactive
toward the carbene complex and that the overall reaction lead to
the formation of a cyclobutanone product
[1]. Since
the alkyne and alkene function are tethered in the starting
enyne the products were bicyclic compounds in which one of the
rings was a cyclobutanone.The structure of the product was also
found to depend on the substitution pattern on the alkene.
For most olefins such as that in enyne 109
a bicyclo-[3.2.0]-heptanone product of the type 110
was produced . If
the terminus of the alkene was disubstituted as in enyne 112,
then a bicyclo-[3.1.1]-heptanone of the type 113
was formed [2].The
mechanism of the formation of this product is thought to be loss
of a CO ligand from the carbene complex followed by addition of
the alkyne to give the vinyl carbene complexed intermediate 115.
Carbon monoxide insertion then would give the ketene
complex 116 or 117.
A [2 + 2] cycloaddition of the ketene and the tethered
alkene in 116 would
lead to the bicyclo-[3.2.0]-heptanone 110.
The same process for ketene complex 117
proceeds in a crossed manner to give the bicyclo-[3.1.1]-heptanone
113. This
switch in the regiochemistry of addition of the alkene to the
ketene presumably
occurs because the more electron-rich end of the double-bond is switched
on |
 |
|
going from the alkene in 109
to that in 112. The reaction has been
found to be stereoselective as indicated in the reaction of the
enyne 118, which
gives a 92 : 8 preference for the isomer 119,
and the enyne 121,
which gives 122 with high stereoselectivity.
The source of the stereoselectivity of these reactions is
currently under investigation as well as the applications of
these reactions to the total synthesis of natural products. |
[i]
[1]
Wulff, W. D.; Kaesler, R. W., Organometallics,
1985, 4, 1461.
[ii][2]
Kim, O. K.; Wulff, W. D.;
Jiang, W.; Ball, R. G., J.
Org. Chem., 1993, 58,
5571.
|