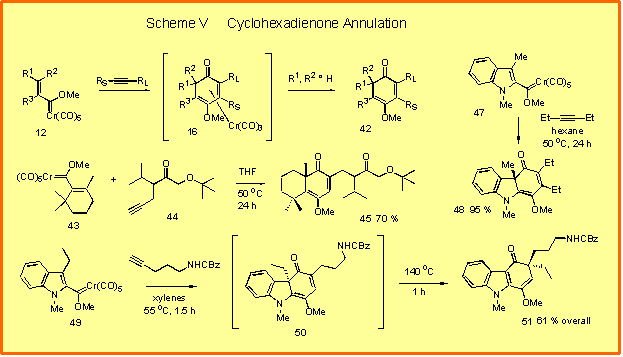
|
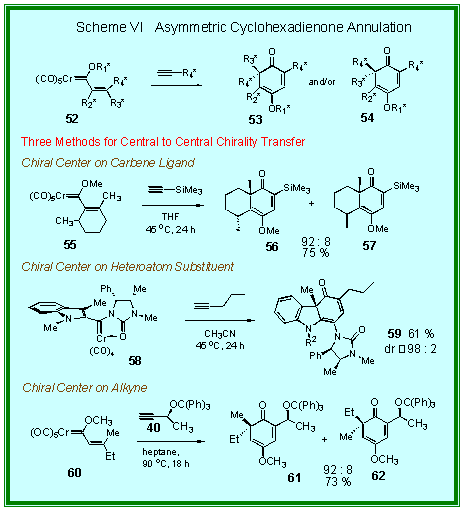
The
only examples of asymmetric induction from chiral heteroatom
substituted carbene complexes involve chiral imidazolidinone
complexes [6].
Complex 58 will react with 1-pentyne to give the cyclohexadienone 59
with greater than 98 : 2 selectivity.
Imidazolidinone complexes of the type 58 exist as isolable atropisomers.
The atropisomer of 58
reacts with 1-pentyne to give the epimer of 59
also with a 98 : 2 selectivity.
As in the benzannulation reaction, the asymmetric
cyclohexadienone annulation occurs with high inductions with
chiral propargylic ethers [7]. The
reaction of the E-carbene complex 60
with alkyne 40 gives a 92 : 8 selectivity for the cyclohexadienone 61
in 73 % yield. This
reaction is also stereospecific since reaction of alkyne 40
with the Z-isomer of carbene complex 60
gives the diastereomer 62
as the major product with similar stereoselectivity ( 91 : 9). This reaction provides for a unique method for 1,4-asymmetric
induction in the construction of highly functionalized
cyclohexadienone and its utilization in total synthesis is
currently ongoing in our research group.
[1]
Tang,
P. C.; Wulff, W. D., J. Am. Chem.
Soc., 1984, 106, 1132.
[2]
Gilbertson, S. R.; Wulff, W. D., Synlett, 1989, 47.
[3]
Bauta, W. E.; Wulff, W. D.; Pavkovic, S. F.; Zaluzec, E. J., J.
Org. Chem., 1989, 54, 3249.
[4]
Quinn,
J. F.; Bos, M. E.; Wulff, W. D., Org.
Lett., 1999, 1,
161.
[5]
Hsung,
R. P.; Wulff, W. D.; Challener, C. A., Synthesis,
1996, 773.
[6]
Quinn,
J. F.; Powers, T. S.; Wulff, W. D.; Yap, G. P. A.; Rheingold, A. L., Organometallics,
1997, 16, 4945.
[7]
Hsung,
R. P.; Quinn, J. F.; Weisenberg, B. A.; Wulff, W. D.; Yap, G. P. A.;
Rheingold, A. L., J. Chem. Soc.,
Chem. Commun., 1997, 615.
|