|
Research Overview The research in our group focuses upon the development and understanding of computational methodologies, and studies in heavy element chemistry, catalysis, protein modeling, drug design/understanding of disease, metal organic frameworks, green chemistry, and many other areas. One of the great features of theoretical and computational chemistry is that they can be utilized to investigate a broad array of challenges, and our group often finds ourselves engaged in areas as diverse as method development and studies of diatomic molecules to protein modeling and studies of the mechanical properties of materials of importance in areas such as aircraft design. Development and understanding of methodologies. Much of our group’s efforts are focused upon the development of ab initio approaches that aim for accurate prediction of thermochemical properties across the periodic table. Included in our efforts has been the development of successful and versatile ab initio composite schemes, called correlation consistent Composite Approaches (ccCA), that provide reduced computational cost (in terms of computer time, memory, and disk space) means to achieve energetic predictions. The approaches are useful for ground-state, excited-state, and transition-state energies, and can be applied to situations where single-reference wavefunctions or where multireference wavefunctions (i.e., bond-breaking, diradicals) are necessary. Included in our work is the development of Gaussian basis sets, providing new additions to the correlation consistent basis set family, and rigorous evaluation of existing and new basis sets. Another area of interest is in gauging the performance of methodologies, such as density functional theory, particularly for situations where there may be few, if any, needed experiments for comparison. Significant efforts have focused upon transition metals.
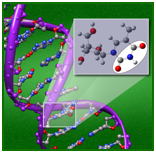
ccCA has been utilized in a broad range of studies, from studies identifying potentially new highly energetic nitrogen species to studies investigating CO2 scrubbers. Here, ccCA was utilized to characterize various possible protonation sites in DNA, sites that may be possible “trigger” sites for the onset of cancer. (from J. Comp. Chem. 2012, 32, 2590; cover article) We have interest in approaches for non-dynamic electron correlation needed to describe bond-breaking and excited states, particularly approaches that could circumvent or reduce the most computationally demanding multireference wavefunction-based approaches. We are identifying diagnostic criteria to assess the need for such approaches for transition metal species, and are developing DFT correction terms to account for non-dynamic electron correlation. Heavy element chemistry. The complexity of the heavy elements results in their great utility in applications from cell phones to stealth technology. We are developing a better understanding of the fundamental properties of lanthanide species, as well as the methodologies needed to describe their energetic and spectroscopic properties, and utilizing this knowledge in areas such as separation science and the development of new methodologies for heavy elements.
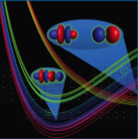
Computed potential energy curves for the neodymium monofluoride dication. Quartet states comprise the lower band of bound curves with the 4I9/2 ground state separated from the excited state manifold by 5 kcal/mol. Homolytic dissociation to NdF2+ and F occurs via crossing to the repulsive sextet states. (from J. Phys. Chem. A 2013, 117, 10881; cover article) Catalysis. Homogeneous and heterogeneous catalysis are of interest, and we investigate a broad range of catalytic reactions, including water-gas shift reactions and reactions of importance in the breakdown of lignin. We also have interest in modeling and trying to improve upon Mother Nature, considering the effectiveness of plant proteins such as RuBisCO, the CO2-fixing enzyme in the Calvin cycle.
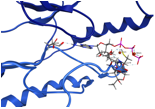
Drug design/understanding disease. In partnership with a pharmaceutical company, we are considering small molecule binding cavities, utilizing docking techniques and other approaches for the design and understanding at the molecular level of potential pharmaceuticals that could be important in anti-inflammatory disease. We also are investigating how changes in structure impact activity, and the role of signal transduction cascades in disease.
|
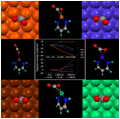
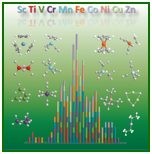 The enthalpies of formation of 225 3d transition metal species were determined using ccCA. This constitutes the largest study of transition metal species done, and these energies can serve as a gauge for more approximate approaches. The figure represents the errors with respect to experiment, and each color represents a different transition metal center (from J. Phys. Chem. A 2012, 116, 870; cover article). A study of 180 4d transition metal species has also been done. (J. Phys. Chem. A 2015, 119, 6867; cover article)
The enthalpies of formation of 225 3d transition metal species were determined using ccCA. This constitutes the largest study of transition metal species done, and these energies can serve as a gauge for more approximate approaches. The figure represents the errors with respect to experiment, and each color represents a different transition metal center (from J. Phys. Chem. A 2012, 116, 870; cover article). A study of 180 4d transition metal species has also been done. (J. Phys. Chem. A 2015, 119, 6867; cover article)