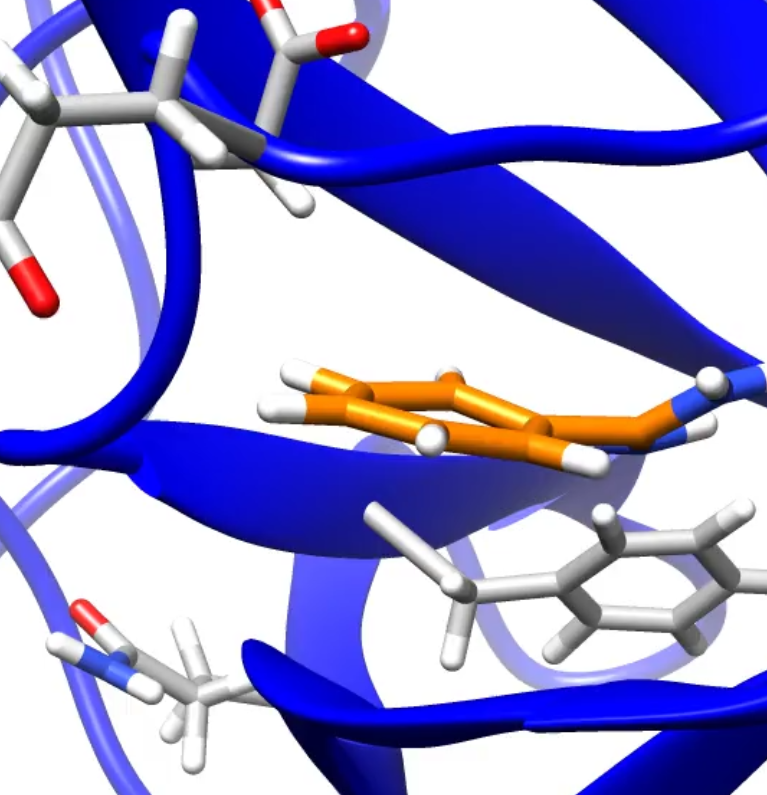
The Per-and Polyfluoroalkyl substances are an important issue for the environment as they can not degrade easily. Moreover, most of the properties for these molecules and their derivatives are unknown, and so need to be assessed. In this project, molecular dynamic is used to study the binding site of different PFAS on different protein present in the Great Lakes fishes as well as on human proteins. Furthermore, quantum molecular calculations are carried on to investigate the different properties of the PFAS molecules (pKa, LogS, etc.).