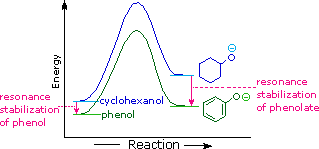
The remarkable stability of the unsaturated hydrocarbon benzene has been discussed in an earlier chapter. The chemical reactivity of benzene contrasts with that of the alkenes in that substitution reactions occur in preference to addition reactions, as illustrated in the following diagram (some comparable reactions of cyclohexene are shown in the green box).
Many other substitution reactions of benzene have been observed, the five most useful are listed below (chlorination and bromination are the most common halogenation reactions). Since the reagents and conditions employed in these reactions are electrophilic, these reactions are commonly referred to as Electrophilic Aromatic Substitution. The catalysts and co-reagents serve to generate the strong electrophilic species needed to effect the initial step of the substitution. The specific electrophile believed to function in each type of reaction is listed in the right hand column.
This mechanism for electrophilic aromatic substitution should be considered in context with other mechanisms involving carbocation intermediates. These include SN1 and E1 reactions of alkyl halides, and Brønsted acid addition reactions to alkenes.
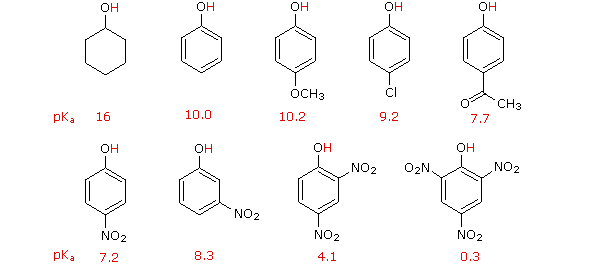
I. The first is the relative reactivity of the compound compared with benzene itself. Experiments have shown that substituents on a benzene ring can influence reactivity in a profound manner.
For example, a hydroxy or methoxy substituent increases the rate of electrophilic substitution about ten thousand fold, as illustrated by the case of anisole in the virtual demonstration (above). In contrast, a nitro substituent decreases the ring's reactivity by roughly a million. This activation or deactivation of the benzene ring toward electrophilic substitution may be correlated with the electron donating or electron withdrawing influence of the substituents, as measured by molecular dipole moments. In the following diagram we see that electron donating substituents (blue dipoles) activate the benzene ring toward electrophilic attack, and electron withdrawing substituents (red dipoles) deactivate the ring (make it less reactive to electrophilic attack).
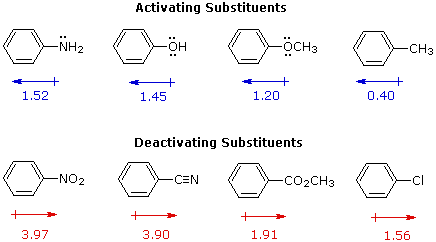 |
The influence a substituent exerts on the reactivity of a benzene ring may be explained by the interaction of two effects:
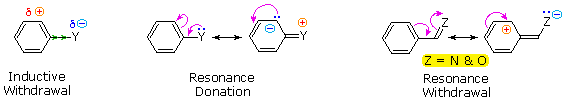
The first is the inductive effect of the substituent. Most elements other than metals and carbon have a significantly greater electronegativity than hydrogen. Consequently, substituents in which nitrogen, oxygen and halogen atoms form sigma-bonds to the aromatic ring exert an inductive electron withdrawal, which deactivates the ring (left-hand diagram below).
The second effect is the result of conjugation of a substituent function with the aromatic ring. This conjugative interaction facilitates electron pair donation or withdrawal, to or from the benzene ring, in a manner different from the inductive shift. If the atom bonded to the ring has one or more non-bonding valence shell electron pairs, as do nitrogen, oxygen and the halogens, electrons may flow into the aromatic ring by p-π conjugation (resonance), as in the middle diagram. Finally, polar double and triple bonds conjugated with the benzene ring may withdraw electrons, as in the right-hand diagram. Note that in the resonance examples all the contributors are not shown. In both cases the charge distribution in the benzene ring is greatest at sites ortho and para to the substituent.
In the case of the nitrogen and oxygen activating groups displayed in the top row of the previous diagram, electron donation by resonance dominates the inductive effect and these compounds show exceptional reactivity in electrophilic substitution reactions. Although halogen atoms have non-bonding valence electron pairs that participate in p-π conjugation, their strong inductive effect predominates, and compounds such as chlorobenzene are less reactive than benzene. The three examples on the left of the bottom row (in the same diagram) are examples of electron withdrawal by conjugation to polar double or triple bonds, and in these cases the inductive effect further enhances the deactivation of the benzene ring. Alkyl substituents such as methyl increase the nucleophilicity of aromatic rings in the same fashion as they act on double bonds.
 |
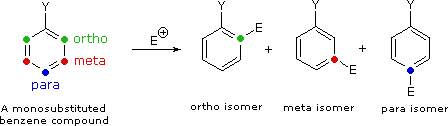
II. The second factor that becomes important in reactions of substituted benzenes concerns the site at which electrophilic substitution occurs. Since a mono-substituted benzene ring has two equivalent ortho-sites, two equivalent meta-sites and a unique para-site, three possible constitutional isomers may be formed in such a substitution. If reaction occurs equally well at all available sites, the expected statistical mixture of isomeric products would be 40% ortho, 40% meta and 20% para. Again we find that the nature of the substituent influences this product ratio in a dramatic fashion. Bromination of methoxybenzene (anisole) is very fast and gives mainly the para-bromo isomer, accompanied by 10% of the ortho-isomer and only a trace of the meta-isomer. Bromination of nitrobenzene requires strong heating and produces the meta-bromo isomer as the chief product.
 |
Some additional examples of product isomer distribution in other electrophilic substitutions are given in the table below. It is important to note here that the reaction conditions for these substitution reactions are not the same, and must be adjusted to fit the reactivity of the reactant C6H5-Y. The high reactivity of anisole, for example, requires that the first two reactions be conducted under very mild conditions (low temperature and little or no catalyst). The nitrobenzene reactant in the third example is very unreactive, so rather harsh reaction conditions must be used to accomplish that reaction.
Y in C6H5–Y | Reaction | % Ortho-Product | % Meta-Product | % Para-Product |
|---|
| –O–CH3 |
Nitration | 30–40 | 0–2 | 60–70 |
| –O–CH3 | F-C Acylation | 5–10 | 0–5 | 90–95 |
| –NO2 | Nitration | 5–8 | 90–95 | 0–5 |
| –CH3 | Nitration | 55–65 | 1–5 | 35–45 |
| –CH3 | Sulfonation | 30–35 | 5–10 | 60–65 |
| –CH3 | F-C Acylation | 10–15 | 2–8 | 85–90 |
| –Br | Nitration | 35–45 | 0–4 | 55–65 |
| –Br | Chlorination | 40–45 | 5–10 | 50–60 |
These observations, and many others like them, have led chemists to formulate an empirical classification of the various substituent groups commonly encountered in aromatic substitution reactions. Thus, substituents that activate the benzene ring toward electrophilic attack generally direct substitution to the ortho and para locations. With some exceptions, such as the halogens, deactivating substituents direct substitution to the meta location. The following table summarizes this classification.
Orientation and Reactivity Effects of Ring Substituents |
|---|
Activating Substituents
ortho & para-Orientation | |
Deactivating Substituents
meta-Orientation | |
Deactivating Substituents
ortho & para-Orientation |
|---|
–O(–)
–OH
–OR
–OC6H5
–OCOCH3 | –NH2
–NR2
–NHCOCH3
–R
–C6H5 | |
–NO2
–NR3(+)
–PR3(+)
–SR2(+)
–SO3H
–SO2R |
–CO2H
–CO2R
–CONH2
–CHO
–COR
–CN |
| |
–F
–Cl
–Br
–I
–CH2Cl
–CH=CHNO2
|
The information summarized in the above table is very useful for rationalizing and predicting the course of aromatic substitution reactions, but in practice most chemists find it desirable to understand the underlying physical principles that contribute to this empirical classification. We have already analyzed the activating or deactivating properties of substituents in terms of inductive and resonance effects, and these same factors may be used to rationalize their influence on substitution orientation.
The first thing to recognize is that the proportions of ortho, meta and para substitution in a given case reflect the relative rates of substitution at each of these sites. If we use the nitration of benzene as a reference, we can assign the rate of reaction at one of the carbons to be 1.0. Since there are six equivalent carbons in benzene, the total rate would be 6.0. If we examine the nitration of toluene, tert-butylbenzene, chlorobenzene and ethyl benzoate in the same manner, we can assign relative rates to the ortho, meta and para sites in each of these compounds. These relative rates are shown (colored red) in the following illustration, and the total rate given below each structure reflects the 2 to 1 ratio of ortho and meta sites to the para position. The overall relative rates of reaction, referenced to benzene as 1.0, are calculated by dividing by six. Clearly, the alkyl substituents activate the benzene ring in the nitration reaction, and the chlorine and ester substituents deactivate the ring.
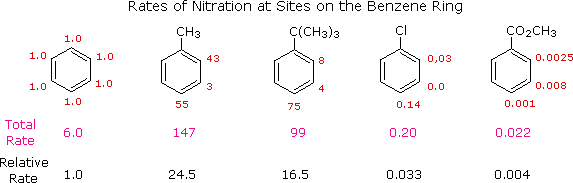 |
From rate data of this kind, it is a simple matter to calculate the proportions of the three substitution isomers. Toluene gives 58.5% ortho-nitrotoluene, 37% para-nitrotoluene and only 4.5% of the meta isomer. The increased bulk of the tert-butyl group hinders attack at the ortho-sites, the overall product mixture being 16% ortho, 8% meta and 75% para-nitro product. Although chlorobenzene is much less reactive than benzene, the rate of ortho and para-substitution greatly exceeds that of meta-substitution, giving a product mixture of 30% ortho and 70% para-nitrochlorobenzene. Finally, the benzoic ester gave predominantly the meta-nitro product (73%) accompanied by the ortho (22%) and para (5%) isomers, as shown by the relative rates. Equivalent rate and product studies for other substitution reactions lead to similar conclusions. For example, electrophilic chlorination of toluene occurs hundreds of times faster than chlorination of benzene, but the relative rates are such that the products are 60% ortho-chlorotoluene, 39% para and 1% meta-isomers, a ratio similar to that observed for nitration.
The manner in which specific substituents influence the orientation of electrophilic substitution of a benzene ring is shown in the following interactive diagram. As noted on the opening illustration, the product-determining step in the substitution mechanism is the first step, which is also the slow or rate determining step. It is not surprising, therefore, that there is a rough correlation between the rate-enhancing effect of a substituent and its site directing influence. The exact influence of a given substituent is best seen by looking at its interactions with the delocalized positive charge on the benzenonium intermediates generated by bonding to the electrophile at each of the three substitution sites. This can be done for seven representative substituents by using the selection buttons underneath the diagram.
In the case of alkyl substituents, charge stabilization is greatest when the alkyl group is bonded to one of the positively charged carbons of the benzenonium intermediate. This happens only for ortho and para electrophilic attack, so such substituents favor formation of those products. Interestingly, primary alkyl substituents, especially methyl, provide greater stabilization of an adjacent charge than do more substituted groups (note the greater reactivity of toluene compared with tert-butylbenzene).
Nitro (NO2), sulfonic acid (SO3H) and carbonyl (C=O) substituents have a full or partial positive charge on the atom bonded to the aromatic ring. Structures in which like-charges are close to each other are destabilized by charge repulsion, so these substituents inhibit ortho and para substitution more than meta substitution. Consequently, meta-products preominate when electrophilic substitution is forced to occur.
Halogen ( X ), OR and NR2 substituents all exert a destabilizing inductive effect on an adjacent positive charge, due to the high electronegativity of the substituent atoms. By itself, this would favor meta-substitution; however, these substituent atoms all have non-bonding valence electron pairs which serve to stabilize an adjacent positive charge by pi-bonding, with resulting delocalization of charge. Consequently, all these substituents direct substitution to ortho and para sites. The balance between inductive electron withdrawal and p-π conjugation is such that the nitrogen and oxygen substituents have an overall stabilizing influence on the benzenonium intermediate and increase the rate of substitution markedly; whereas halogen substituents have an overall destabilizing influence.
3. Characteristics of Specific Substitution Reactions
| Halogenation: |
C6H6 | + Cl2 & heat
FeCl3 catalyst | ——> | C6H5Cl
Chlorobenzene | + HCl |
| Nitration: |
C6H6 | + HNO3 & heat
H2SO4 catalyst | ——> | C6H5NO2
Nitrobenzene |
+ H2O |
| Sulfonation: |
C6H6 | + H2SO4 + SO3
& heat | ——> | C6H5SO3H
Benzenesulfonic acid |
+ H2O |
Alkylation:
Friedel-Crafts |
C6H6 | + R-Cl & heat
AlCl3 catalyst | ——> | C6H5-R
An Arene |
+ HCl |
Acylation:
Friedel-Crafts |
C6H6 | + RCOCl & heat
AlCl3 catalyst | ——> | C6H5COR
An Aryl Ketone |
+ HCl |
|
The conditions commonly used for the aromatic substitution reactions discussed here are repeated in the table on the right. The electrophilic reactivity of these different reagents varies. We find, for example, that nitration of nitrobenzene occurs smoothly at 95 ºC, giving meta-dinitrobenzene, whereas bromination of nitrobenzene (ferric catalyst) requires a temperature of 140 ºC. Also, as noted earlier, toluene undergoes nitration about 25 times faster than benzene, but chlorination of toluene is over 500 times faster than that of benzene. From this we may conclude that the nitration reagent is more reactive and less selective than the halogenation reagents.
Both sulfonation and nitration yield water as a by-product. This does not significantly affect the nitration reaction (note the presence of sulfuric acid as a dehydrating agent), but sulfonation is reversible and is driven to completion by addition of sulfur trioxide, which converts the water to sulfuric acid. The reversibility of the sulfonation reaction is occasionally useful for removing this functional group.
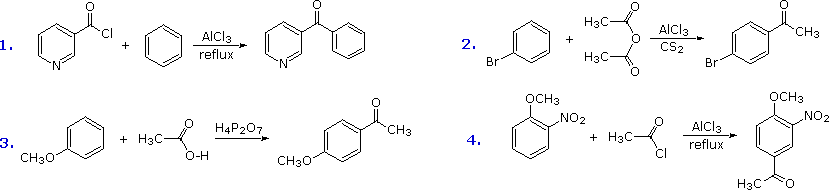
The Friedel-Crafts acylation reagent is normally composed of an acyl halide or anhydride mixed with a Lewis acid catalyst such as AlCl3. This produces an acylium cation, R-C≡O(+), or a related species. Such electrophiles are not exceptionally reactive, so the acylation reaction is generally restricted to aromatic systems that are at least as reactive as chlorobenzene. Carbon disulfide is often used as a solvent, since it is unreactive and is easily removed from the product. If the substrate is a very reactive benzene derivative, such as anisole, carboxylic esters or acids may be the source of the acylating electrophile. Some examples of Friedel-Crafts acylation reactions are shown in the following diagram. The first demonstrates that unusual acylating agents may be used as reactants. The second makes use of an anhydride acylating reagent, and the third illustrates the ease with which anisole reacts, as noted earlier. The H4P2O7 reagent used here is an anhydride of phosphoric acid called pyrophosphoric acid. Finally, the fourth example illustrates several important points. Since the nitro group is a powerful deactivating substituent, Friedel-Crafts acylation of nitrobenzene does not take place under any conditions. However, the presence of a second strongly-activating substituent group permits acylation; the site of reaction is that favored by both substituents.
A common characteristic of the halogenation, nitration, sulfonation and acylation reactions is that they introduce a deactivating substituent on the benzene ring. As a result, we do not normally have to worry about disubstitution products being formed. Friedel-Crafts alkylation, on the other hand, introduces an activating substituent (an alkyl group), so more than one substitution may take place. If benzene is to be alkylated, as in the following synthesis of tert-butylbenzene, the mono-alkylated product is favored by using a large excess of this reactant. When the molar ratio of benzene to alkyl halide falls below 1:1, para-ditert-butylbenzene becomes the major product.
C6H6 (large excess) + (CH3)3C-Cl + AlCl3 ——> C6H5-C(CH3)3 + HCl
The carbocation electrophiles required for alkylation may be generated from alkyl halides (as above), alkenes + strong acid or alcohols + strong acid. Since 1º-carbocations are prone to rearrangement, it is usually not possible to introduce 1º-alkyl substituents larger than ethyl by Friedel-Crafts alkylation. For example, reaction of excess benzene with 1-chloropropane and aluminum chloride gives a good yield of isopropylbenzene (cumene).
C6H6 (large excess) + CH3CH2CH2-Cl + AlCl3 ——> C6H5-CH(CH3)2 + HCl
Additional examples of Friedel-Crafts alkylation reactions are shown in the following diagram.
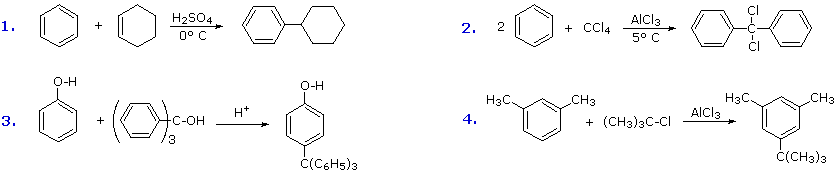
The first and third examples show how alkenes and alcohols may be the source of the electrophilic carbocation reactant. The triphenylmethyl cation generated in the third case is relatively unreactive, due to extensive resonance charge delocalization, and only substitutes highly activated aromatic rings. The second example shows an interesting case in which a polychlororeactant is used as the alkylating agent. A four fold excess of carbon tetrachloride is used to avoid tri-alkylation of this reagent, a process that is retarded by steric hindrance. The fourth example illustrates the poor orientational selectivity often found in alkylation reactions of activated benzene rings. The bulky tert-butyl group ends up attached to the reactive meta-xylene ring at the least hindered site. This may not be the site of initial bonding, since polyalkylbenzenes rearrange under Friedel-Crafts conditions (para-dipropylbenzene rearranges to meta-dipropylbenzene on heating with AlCl3).
A practical concern in the use of electrophilic aromatic substitution reactions in synthesis is the separation of isomer mixtures. This is particularly true for cases of ortho-para substitution, which often produce significant amounts of the minor isomer. As a rule, para-isomers predominate except for some reactions of toluene and related alkyl benzenes. Separation of these mixtures is aided by the fact that para-isomers have significantly higher melting points than their ortho counterparts; consequently, fractional crystallization is often an effective isolation technique. Since meta-substitution favors a single product, separation of trace isomers is normally not a problem.
4. Electrophilic Substitution of Disubstituted Benzene Rings
When a benzene ring has two substituent groups, each exerts an influence on subsequent substitution reactions. The activation or deactivation of the ring can be predicted more or less by the sum of the individual effects of these substituents. The site at which a new substituent is introduced depends on the orientation of the existing groups and their individual directing effects. We can identify two general behavior categories, as shown in the following table. Thus, the groups may be oriented in such a manner that their directing influences act in concert, reinforcing the outcome; or are opposed (antagonistic) to each other. Note that the orientations in each category change depending on whether the groups have similar or opposite individual directing effects.
Orientational Interaction of Substituents |
|---|
Antagonistic or Non-Cooperative | | Reinforcing or Cooperative |
|---|
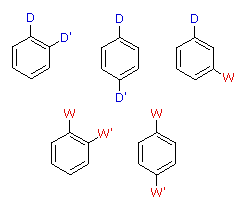 | | 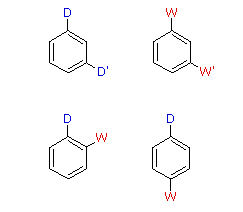
| D = Electron Donating Group (ortho/para-directing)
W = Electron Withdrawing Group (meta-directing) | |
The products from substitution reactions of compounds having a reinforcing orientation of substituents are easier to predict than those having antagonistic substituents. For example, the six equations shown below are all examples of reinforcing or cooperative directing effects operating in the expected manner. Symmetry, as in the first two cases, makes it easy to predict the site at which substitution is likely to occur. Note that if two different sites are favored, substitution will usually occur at the one that is least hindered by ortho groups.
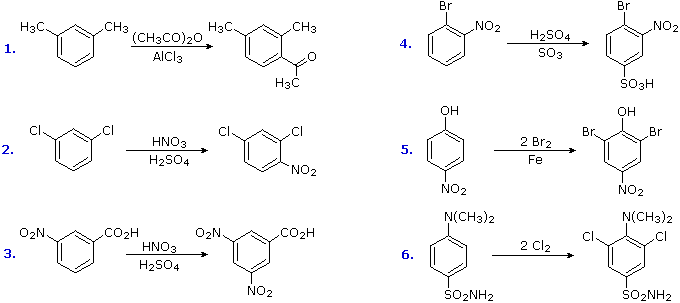
The first three examples have two similar directing groups in a meta-relationship to each other. In examples 4 through 6, oppositely directing groups have an ortho or para-relationship. The major products of electrophilic substitution, as shown, are the sum of the individual group effects. The strongly activating hydroxyl (–OH) and amino (–NH2) substituents favor dihalogenation in examples 5 and six.
Substitution reactions of compounds having an antagonistic orientation of substituents require a more careful analysis. If the substituents are identical, as in example 1 below, the symmetry of the molecule will again simplify the decision. When one substituent has a pair of non-bonding electrons available for adjacent charge stabilization, it will normally exert the product determining influence, examples 2, 4 & 5, even though it may be overall deactivating (case 2). Case 3 reflects a combination of steric hindrance and the superior innate stabilizing ability of methyl groups relative to other alkyl substituents. Example 6 is interesting in that it demonstrates the conversion of an activating ortho/para-directing group into a deactivating meta-directing "onium" cation [–NH(CH3)2(+) ] in a strong acid environment.
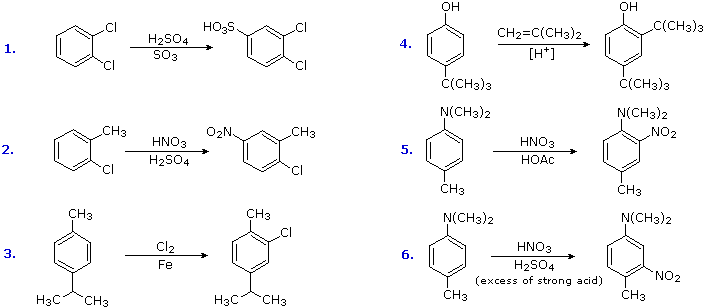

Reactions of Substituent Groups
1. Oxidation of Alkyl Side-Chains
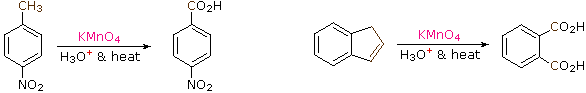
The benzylic hydrogens of alkyl substituents on a benzene ring are activated toward free radical attack, as noted earlier. Furthermore, SN1, SN2 and E1 reactions of benzylic halides, show enhanced reactivity, due to the adjacent aromatic ring. The possibility that these observations reflect a general benzylic activation is supported by the susceptability of alkyl side-chains to oxidative degradation, as shown in the following examples (the oxidized side chain is colored). Such oxidations are normally effected by hot acidic pemanganate solutions, but for large scale industrial operations catalysed air-oxidations are preferred. Interstingly, if the benzylic position is completely substituted this oxidative degradation does not occur (second equation, the substituted benzylic carbon is colored blue).
| C6H5–CH2CH2CH2CH3 + KMnO4 + H3O(+) & heat |  | C6H5–CO2H + CO2 |
| p-(CH3)3C–C6H4–CH3 + KMnO4 + H3O(+) & heat | | p-(CH3)3C–C6H4–CO2H |
These equations are not balanced. The permanganate oxidant is reduced, usually to Mn(IV) or Mn(II). Two other examples of this reaction are given below, and illustrate its usefulness in preparing substituted benzoic acids.
2. Reduction of Nitro Groups and Aryl Ketones
Electrophilic nitration and Friedel-Crafts acylation reactions introduce deactivating, meta-directing substituents on an aromatic ring. The attached atoms are in a high oxidation state, and their reduction converts these electron withdrawing functions into electron donating amino and alkyl groups. Reduction is easily achieved either by catalytic hydrogenation (H2 + catalyst), or with reducing metals in acid. Examples of these reductions are shown here, equation 6 demonstrating the simultaneous reduction of both functions. Note that the butylbenzene product in equation 4 cannot be generated by direct Friedel-Crafts alkylation due to carbocation rearrangement. The zinc used in ketone reductions, such as 5, is usually activated by alloying with mercury (a process known as amalgamation).
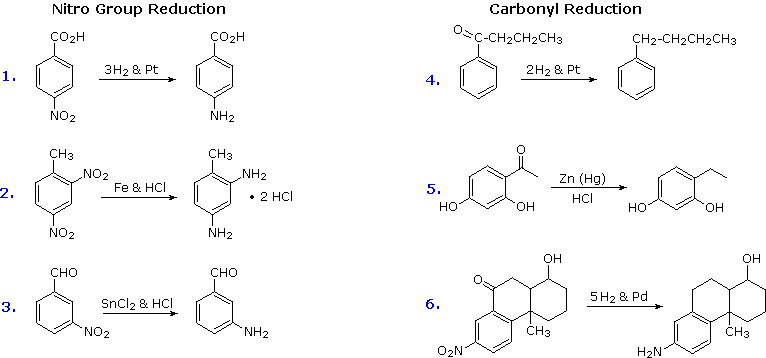
Several alternative methods for reducing nitro groups to amines are known. These include zinc or tin in dilute mineral acid, and sodium sulfide in ammonium hydroxide solution. The procedures described above are sufficient for most cases.
3. Conversion of Halogens to Organometallic Reagents
The reaction of alkyl and aryl halides with reactive metals (usually Li & Mg) to give nucleophilic reagents has been noted. This provides a powerful tool for the conversion of chloro, bromo or iodo substituents into a variety of other groups. Many reactions of these aryl lithium and Grignard reagents will be discussed in later sections, and the following equations provide typical examples of carboxylation, protonation and Gilman coupling. Metal halogen exchange reactions take place at low temperature, and may be used to introduce iodine at designated locations. An example of this method will be displayed below by clicking on the diagram. In this example care must be taken to maintain a low temperature, because elimination to an aryne intermediate takes place on warming.
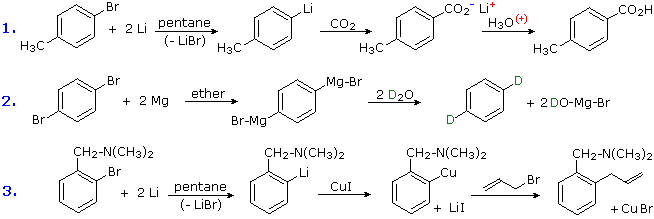
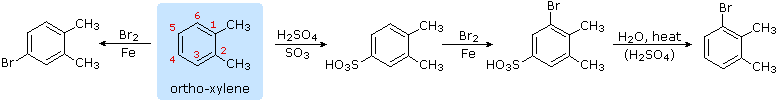
4. Hydrolysis of Sulfonic Acids
The potential reversibility of the aromatic sulfonation reaction was noted earlier. The following equation illustrates how this characteristic of the sulfonic acids may be used to prepare the 3-bromo derivative of ortho-xylene. Direct bromination would give the 4-bromo derivative.
5. Modifying the Influence of Strong Activating Groups
The strongest activating and ortho/para-directing substituents are the amino (-NH2) and hydroxyl (-OH) groups. Direct nitration of phenol (hydroxybenzene) by dilute nitric acid gives modest yields of nitrated phenols and considerable oxidative decomposition to tarry materials; aniline (aminobenzene) is largely destroyed. Bromination of both phenol and aniline is difficult to control, with di- and tri-bromo products forming readily. Because of their high nucleophilic reactivity, aniline and phenol undergo substitution reactions with iodine, a halogen that is normally unreactive with benzene derivatives. The mixed halogen iodine chloride (ICl) provides a more electrophilic iodine moiety, and is effective in iodinating aromatic rings having less powerful activating substituents.
| C6H5–NH2 + I2 + NaHCO3 | | | p-I–C6H4–NH2 + NaI + CO2 + H2O |
By acetylating the heteroatom substituent on phenol and aniline, its activating influence can be substantially attenuated. For example, acetylation of aniline gives acetanilide (first step in the following equation), which undergoes nitration at low temperature, yielding the para-nitro product in high yield. The modifying acetyl group can then be removed by acid-catalyzed hydrolysis (last step), to yield para-nitroaniline. Although the activating influence of the amino group has been reduced by this procedure, the acetyl derivative remains an ortho/para-directing and activating substituent.
C6H5–NH2 + (CH3CO)2O |
pyridine (a base)
|
C6H5–NHCOCH3 | HNO3 , 5 ºC
 |
p-O2N–C6H4–NHCOCH3 | H3O(+) & heat
|
p-O2N–C6H4–NH2 |
The following diagram illustrates how the acetyl group acts to attenuate the overall electron donating character of oxygen and nitrogen. The non-bonding valence electron pairs that are responsible for the high reactivity of these compounds (blue arrows) are diverted to the adjacent carbonyl group (green arrows). However, the overall influence of the modified substituent is still activating and ortho/para-directing.
|
It should now be apparent that an extensive "toolchest" of reactions are available to us for the synthesis of substituted benzenes. Just as an expert carpenter must understand the characteristics and limitations of his/her tools, chemists must appreciate the nature of their "tools" when applying them to a specific synthesis. Six proposed syntheses are listed in the following diagram in rough order of increasing complexity. You should try to conceive a plausible reaction sequence for each. Once you have done so, you may check suggested answers by clicking on the question mark for each.
|
Reactions of Fused Benzene Rings
Compounds in which two or more benzene rings are fused together were described in an earlier chapter, and they present interesting insights into aromaticity and reactivity. The smallest such hydrocarbon is naphthalene. Naphthalene is stabilized by resonance. Three canonical resonance contributors may be drawn, and are displayed in the following diagram.
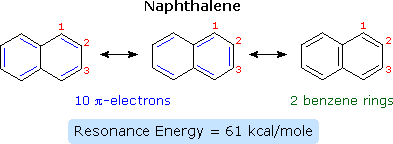
The two structures on the left have one discrete benzene ring each, but may also be viewed as 10-pi-electron annulenes having a bridging single bond. The structure on the right has two benzene rings which share a common double bond. From heats of hydrogenation or combustion, the resonance energy of naphthalene is calculated to be 61 kcal/mole, 11 kcal/mole less than that of two benzene rings (2 * 36). As expected from an average of the three resonance contributors, the carbon-carbon bonds in naphthalene show variation in length, suggesting some localization of the double bonds. The C1–C2 bond is 1.36 Å long, whereas the C2–C3 bond length is 1.42 Å. This contrasts with the structure of benzene, in which all the C–C bonds have a common length, 1.39 Å.
Naphthalene is more reactive than benzene, both in substitution and addition reactions, and these reactions tend to proceed in a manner that maintains one intact benzene ring. The following diagram shows three oxidation and reduction reactions that illustrate this feature. In the last example, catalytic hydrogenation of one ring takes place under milder conditions than those required for complete saturation (the decalin product exists as cis/trans isomers). Electrophilic substitution reactions take place more rapidly at C1, although the C2 product is more stable and predominates at equilibrium. Examples of these reactions will be displayed by clicking on the diagram. The kinetically favored C1 orientation reflects a preference for generating a cationic intermediate that maintains one intact benzene ring. By clicking on the diagram a second time, the two naphthenonium intermediates created by attack at C1 and C2 will be displayed.
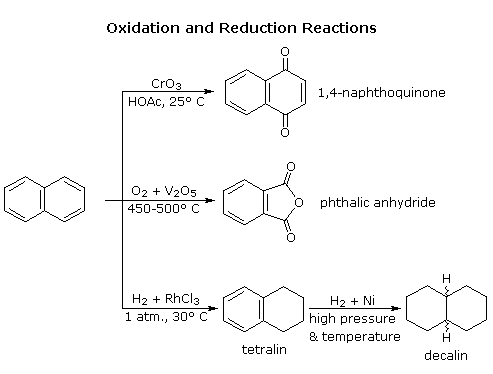
The structure and chemistry of more highly fused benzene ring compounds, such as anthracene and phenanthrene show many of the same characteristics described above.
Nucleophilic Substitution, Elimination & Addition Reactions of Benzene Derivatives
1. Substitution
An early method of preparing phenol (the Dow process) involved the reaction of chlorobenzene with a concentrated sodium hydroxide solution at temperatures above 350 ºC. The chief products are phenol and diphenyl ether (see below). This apparent nucleophilic substitution reaction is surprising, since aryl halides are generally incapable of reacting by either an SN1 or SN2 pathway.
C6H5–Cl + NaOH solution |
350 ºC
|
C6H5–OH + C6H5–O–C6H5 + NaCl |
The presence of electron-withdrawing groups (such as nitro) ortho and para to the chlorine substancially enhance the rate of substitution, as shown in the set of equations presented on the left below. To explain this, a third mechanism for nucleophilic substitution has been proposed. This two-step mechanism is characterized by initial addition of the nucleophile (hydroxide ion or water) to the aromatic ring, followed by loss of a halide anion from the negatively charged intermediate. This is illustrated by clicking the "Show Mechanism" button next to the diagram. The sites over which the negative charge is delocalized are colored blue, and the ability of nitro, and other electron withdrawing, groups to stabilize adjacent negative charge accounts for their rate enhancing influence at the ortho and para locations.
Three additional examples of aryl halide nucleophilic substitution are presented on the right. Only the 2- and 4-chloropyridine isomers undergo rapid substitution, the 3-chloro isomer is relatively unreactive. Nitrogen nucleophiles will also react, as evidenced by the use of Sanger's reagent for the derivatization of amino acids. The resulting N-2,4-dinitrophenyl derivatives are bright yellow crystalline compounds that facilitated analysis of peptides and proteins, a subject for which Frederick Sanger received one of his two Nobel Prizes in chemistry.
Such addition-elimination processes generally occur at sp2 or sp hybridized carbon atoms, in contrast to SN1 and SN2 reactions. When applied to aromatic halides, as in the present discussion, this mechanism is called SNAr. Some distinguishing features of the three common nucleophilic substitution mechanisms are summarized in the following table.
Mechanism | Number of Steps | Bond Formation Timing | Carbon Hybridization |
|---|
SN1 | Two | After Bond Breaking | Usually sp3 |
|---|
SN2 | One | Simultaneous with
Bond Breaking | Usually sp3 |
|---|
SNAr | Two | Prior to Bond Breaking | Usually sp2 |
|---|
2. Elimination
There is good evidence that the synthesis of phenol from chlorobenzene does not proceed by the addition-elimination mechanism (SNAr) described above. For example, treatment of para-chlorotoluene with sodium hydroxide solution at temperatures above 350 ºC gave an equimolar mixture of meta- and para-cresols (hydroxytoluenes). Chloro and bromobenzene reacted with the very strong base sodium amide (NaNH2 at low temperature (-33 ºC in liquid ammonia) to give good yields of aniline (aminobenzene). However, ortho-chloroanisole gave exclusively meta-methoxyaniline under the same conditions. These reactions are described by the following equations.
The explanation for this curious repositioning of the substituent group lies in a different two-step mechanism we can refer to as an elimination-addition process. The intermediate in this mechanism is an unstable benzyne species, as displayed in the above illustration by clicking the "Show Mechanism" button. In contrast to the parallel overlap of p-orbitals in a stable alkyne triple bond, the p-orbitals of a benzyne are tilted ca.120º apart, so the reactivity of this incipient triple bond to addition reactions is greatly enhanced. In the absence of steric hindrance (top example) equal amounts of meta- and para-cresols are obtained. The steric bulk of the methoxy group and the ability of its ether oxygen to stabilize an adjacent anion result in a substantial bias in the addition of amide anion or ammonia.
|
For additional information about benzyne and related species , Click Here. |
3. Addition
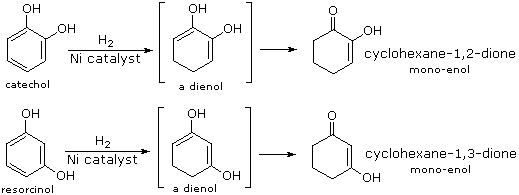
Although it does so less readily than simple alkenes or dienes, benzene adds hydrogen at high pressure in the presence of Pt, Pd or Ni catalysts. The product is cyclohexane and the heat of reaction provides evidence of benzene's thermodynamic stability. Substituted benzene rings may also be deduced in this fashion, and hydroxy-substituted compounds, such as phenol, catechol and resorcinol, give carbonyl products resulting from the fast ketonization of intermediate enols. Nickel catalysts are often used for this purpose, as noted in the following equations.
Benzene is more susceptible to radical addition reactions than to electrophilic addition. We have already noted that benzene does not react with chlorine or bromine in the absence of a catalyst and heat. In strong sunlight or with radical initiators benzene adds these halogens to give hexahalocyclohexanes. It is worth noting that these same conditions effect radical substitution of cyclohexane, the key factors in this change of behavior are the pi-bonds array in benzene, which permit addition, and the weaker C-H bonds in cyclohexane. The addition of chlorine is shown below; two of the seven meso-stereoisomers will appear if the "Show Isomer" button is clicked.